

8209
.pdfЭнергия активации процесса диссоциации также довольно велика (для MgCO3 -110-
140 кДж/моль). Поскольку, чем выше энергия активации процесса, тем сильнее его скорость зависит от температуры, поэтому скорость разложения карбонатов очень сильно зависит от температуры. Например, для кальцита повышение температуры сверх температуры диссоциации на каждые 100°C ускорят декарбонизацию примерно в 30 раз.
Дегидратация
Минералы многих сырьевых материалов содержат в своем составе связанную воду. Конечно, сырье обладает и естественной влажностью, то есть содержит свободную воду в капельно-жидком состоянии, но далее речь пойдет именно о связанной воде.
В зависимости от вида и роли воды в структуре ее можно разделить на
Конституционную,
Кристаллизационную,
Адсорбционную.
При этом конституционная и кристаллизационная относится к химически связанной, а адсорбционная – к физически связанной воде.
Конституционная вода входит в структуру соединений в виде аниона ОН- и реже иона гидроксония Н3О+, которые являются элементами строения кристаллической решетки. Соединения, содержащие ОН- называются гидроксидами. Удаление такой воды сопровождается полным разрушением структуры исходного соединения и образованием оксида с совершенно иной структурой.
Кристаллизационная вода входит в состав минералов в виде молекул Н2О, а сами вещества называются гидратами. При этом, гидраты могут иметь в своем составе либо строго постоянное содержание молекул воды (кристаллогидраты), либо ее содержание может варьироваться.
Вкристаллогидратах молекулярная вода входит в состав кристаллической решетки, занимая в ней определенные кристаллографические положения. Удаление такой воды, как и в случае гидроксидов, сопровождается полным разрушением структуры исходного соединения с образованием соединения с иной структурой.
Вгидратах с переменным составом вода не входит в состав кристаллической решетки в качестве структурной единицы, а располагается в межузлиях. пустотах и каналах структуры, хотя образоваться такие вещества могут только в присутствии воды. Такие гидраты условно можно рассматривать как твердые растворы внедрения, равновесное содержание воды в которых определяется температурой и парциальным давлением паров воды. Удаление такой воды не сопровождается разрушением структуры исходного соединения, при чем, при обезвоживании и изменении внешних условий (влажности среды) вещество снова может поглощать воду и она входит в структуру, не изменяя ее строение. Примерами таких гидратов являются молекулярные сита или цеолиты (они относятся к классу алюмосиликатов и содержат цеолитную воду), а также глинистые минералы и кристаллы полуводного гипса.
Адсорбционная (гигроскопическая) вода содержит физически связанную воду,
которая удерживается на поверхности частиц, а также в порах и капиллярах зерен за счет сил межмолекулярного и электростатического взаимодействия. Такая вода образует адсорбционный слой в одну или несколько молекул. При этом сохраняется способность молекул диффундировать вдоль поверхности частиц и удаляться с поверхности за счет
41
теплового движения. Воду, адсорбированную на поверхности отдельных слое в минералах сложной слоистой структуры, называют межслоевой водой (такая встречается в глинистых минералах), а воду, находящуюся в порах и капиллярах, называют,
соответственно, поровой и капиллярной.
Поскольку адсорбция часто сопровождается хемосорбцией, то есть химическим взаимодействием воды со структурой минерала, то иногда бывает довольно сложно провести четкую границу между физически и химически связанной водой. Адсорбционная вода отличается по свойствам (плотности, температуре замерзания и пр.) от капельножидкой воды, так. Капиллярная вода замерзает гораздо ниже 0°С.
Дегидратация – процесс удаления из минералов связанной воды, который происходит под воздействием внешних факторов
Дегидратацию может инициировать нагревание вещества, понижение влажности окружающей среды, взаимодействие с другим веществом, связывающим воду (осушителем). Термин дегидратация обычно применяется в отношении удаления конституционной и кристаллизационной воды, в отношении удаления адсорбционной воды принят термин обезвоживание, а если речь идет об удалении капельно-жидкой воды, то говорят о сушке.
Потеря конституционной воды является частным случаем реакции диссоциации,
при которой вещество теряет воду при достижении определенной температуры, когда давление диссоциации достигает внешнего давления, в результате чего образуется водяной пар и новая твердая фаза. При этом давление диссоциации сильно зависит от температуры. Так дегидратируются гидроксиды Са(ОН)2 и Mg(OH)2. Аналогично дегидратируются некоторые кристаллогидраты типа сингенита K2SO4·CaSO4·H2O.
Есть такие кристаллогидраты, которые при нагревании сначала плавятся в своей кристаллизационной воде. При этом возможны два случая.
1.Кристаллогидрат может плавиться без разложения (конгруентно), образуя насыщенный раствор. При нагревании раствор закипает, и его температура повышается до тех пор, пока раствор не станет насыщенным, после чего он кипит при постоянной температуре, пока не образуется безводный продукт. Так ведет себя гипосульфит натрия
Na2SO4·10H2O
2.Кристаллогидрат может плавиться инконгруентно, образуя безводную соль или гидрат с меньшим содержанием воды и насыщенный по отношению к ним раствор, который выкипает при постоянной температуре. Так дегидратируется мирабилит Na2SO4·10 H2O или медный купорос CuSO4·5H2O
Дегидратация гидрокисдов и кристаллогидратов сопровождается перестройкой структуры исходного твердого вещества и возникновением новой структуры безводного соединения. Однако встречаются минералы, которые после дегидратации сохраняют в течение какого-то времени свою структуру, но при этом состояние в этом случае будет носить метастабильный характер. При изменении условий эта метастабильная фаза может перейти в стабильное состояние с уже иной структурой. Примером этого является дегидратация брусита Mg(OH)2. Для него при 375-380°С потеря воды сначала сопровождается образованием MgO со структурой брусита, которая только при более высокой температуре переходит в стабильную кристаллическую структуру периклаза.
Часто при дегидратации получаются безводные продукты в аморфном состоянии. Например, при дегидратации каолинита Al2O3·2SiO2·2H2O потеря воды при 500-600°С сопровождается разрушением его структуры и образованием аморфного продукта
42
метакаолинита Al2O3·2SiO2 (по другим данным - смеси аморфных Al2O3 и·SiO2 ), который при дальнейшем нагревании превращаются в кристаллический муллит 3Al2O3·2SiO2.
Цеолитная вода удаляется из молекулярных сит без изменений в их структуре. Молекулы воды располагаются внутри соединенных с поверхностью кристалла крупных структурных полостей алюмосиликатного пространственного состава. Энергия связи воды со структурой невелика, и разрушение такой связи не ведет к разрушению каркаса, удаление воды происходит плавно, начинаясь уже при невысоких температурах даже без нагрева, но при условии пониженной влажности среды.
Дегидратация – процесс эндотермический, то есть требует подвода тепла к системе. Теплота разложения, например, Ca(OH)2 при 520°С составляет -123 кДж/моль
Под твердофазовыми реакциями понимают реакции, протекающие за счет непосредственного взаимодействия между частицами твердых веществ без участия жидкой или газовой фазы.
Учение о реакциях в твердом состоянии имеет большое значение для химии и технологии силикатных и других тугоплавких материалов, поскольку в процессе их производства при высокотемпературной обработке исходных сырьевых смесей и сформованных из них изделий взаимодействие составляющих их компонентов начинается именно с твердофазовых реакций.
В общем виде схему твердофазовой реакции можно представить следующим образом: при некоторой температуре, характерной для каждого из реагирующих веществ, на поверхности их контакта амплитуда колебаний атомов в решетка оказывается достаточной для того, чтобы начался процесс обмена местами между отдельными атомами реагирующих веществ, в результата чего начинается химическая реакция, приводящая к образованию продукта реакции — вещества АВ. После образования на поверхности контакта слоя вещества АВ дальнейшее протекание реакции будет возможным только при наличии диффузии вещества А через слой продукта реакции АВ к веществу В и (или) наоборот — диффузии вещества В через слой АВ к веществу А.
Из приведенной схемы твердофазовой реакции следует, что ее скорость может лимитироваться двумя факторами: скоростью химического взаимодействия компонентов и скоростью процесса диффузии. В силикатных и ряде других систем скорость взаимодействия между твердыми фазами на самых ранних стадиях процесса может определяться скоростью химической реакции. Сразу же после образования слоя продукта реакции ее скорость начинает лимитироваться скоростью процесса диффузии, поскольку в большинстве случаев скорость химического взаимодействия превышает скорость диффузии. Поэтому именно процесс диффузии оказывает решающее влияние на ход большинства твердофазовых реакций.
В реальных условиях твердофазовые реакции обычно протекают в порошкообразных смесях. При этом одно из реагирующих веществ называют
покрывающим, а другое — покрываемым компонентом. Покрывающий компонент, как правило, обладает большим коэффициентом диффузии (обычно, но не всегда он имеет более низкую температуру плавления). При реакции частицы покрывающего компонента как бы обволакивают, покрывают зерна покрываемого компонента.
Особенности реакций в твердом состоянии
43
Выделение твердофазовых реакций в особую группу связано с их специфическим характером, во многом отличным от реакций в газах и жидкостях. Среди особенностей твердофазовых реакций можно отметить следующие.
1.Направление химической реакции в смесях твердых веществ может не совпадать
снаправлением реакции в жидкой фазе. Например, в обычных условиях в водных растворах угольная кислота сильнее кремниевой, а при твердофазовом взаимодействии кремниевая кислота вытесняет угольную из ее солей.
2.Процесс взаимодействия твердых компонентов имеет ступенчатый характер. Если в данной системе существует несколько различных соединений, то процесс образования конечного продукта реакции проходит через ряд стадий, заключающихся в последовательном образовании промежуточных продуктов, причем любом соотношении исходных компонентов (т.е. при различном составе конечного продукта реакции) реализуется одна и та же последовательность образования промежуточных соединений. а первично образующейся в данной конкретной системе фазой является одно и то же определенное соединение. Например, в системе CaO—SiО2 при любом молярном соотношении СаО/ SiО2 в исходной смеси вначале всегда образуется ортосиликат кальция
2СаО·SiО2. На рис. 83 изображена, например, последовательность образования соединений в смеси СаО и SiО2 с соотношенем СаО/ SiО2 = 1:1 при 1200°С. Вначале образуется 2СаО·SiО2 количество которого на первой стадии реакции непрерывно увеличивается, достигая максимума, после чего начинает падать за счет взаимодействия с SiО2 и образования конечного продукта реакции — метасиликата кальция СаО·SiО2. Кроме 2СаО·SiО2 в качестве промежуточного продукта в этой системе образуя также соединение 3СаО·2SiО2 .
3. Твердофазовые реакции всегда идут с выделением тепла, т.е. являются экзотермическими. Следует, однако, отметить, что эта особенность относится только к чисто твердофазовым реакциям, в которых не участвует газовая или жидкая фаза. Величина тепловыделения при реакциях твердых веществ достигает значительных величин: например, в силикатных системах она может составлять несколько сотен и более
кДж/моль.
4. Если при твердофазовом взаимодействии не образуются твердые растворы и не возникает значительных количеств газовой и жидкий фаз, то термодинамическоое равновесие в реакции практически не может быть достигнуто, т.е. реакция идет в одном направлении до конца, пока не будет полностью израсходован по крайней мере один из реагирующих компонентов. Равновесие может быть достигнуто при малой теплоте реакции и значительной разнице между суммой теплоемкостей исходных веществ и продуктов реакции, но эти условия в реальных силикатных системах, как правило, не выполняются. Следует, однако, подчеркнуть, что степень завершенности реакции определяется не только термодинамическими, но и чисто кинетическими факторами, поэтому практически твердофазовые реакции могут и не доходить до конца.
Кинетика твердофазовых реакций
Предложено достаточно много зависимостей, описывающих кинетику твердофазовых реакций. Эти зависимости характеризуют изменение во времени количества веществ, участвующих в реакции. Следует, однако, отметить, что получить какое-то общее уравнение для указанной зависимости, пригодное для всей реакции в целом, без ряда допущений и упрощений вряд ли возможно. Это связано с тем, что твердофазовые реакции складываются из ряда стадий, кинетика которых описывается
44
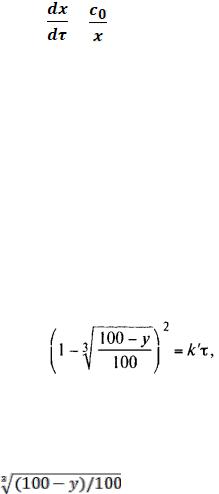
различными закономерностями. Так, вначале скорость твердофазового взаимодействия может определяться скоростью химического взаимодействия между компонентами, а впоследствии — скоростью процесса диффузии, причем в каждом конкретном случае переход из области, контролируемой законами химической кинетики, в область, контролируемую законами диффузионной кинетики, может происходить в различные моменты времени.
Одна из первых формул, описывающих кинетику твердофазовых реакций, было предложена В. Яндером. Уравнение Яндера, выведенное исходя из закона Фика, для стадии твердофазовой реакции, контролируемой процессом диффузии, имеет для изотермических условий вид:
=D |
(1) |
|
Где х — толщина слоя продукта реакции на покрываемом, менее подвижном |
||
компоненте; τ |
— время; D — коэффициент диффузии при данной температуре; с0 — |
|
концентрация диффундирующего компонента на границе со слоем продукта реакции. |
||
Принимая величины D и с0 постоянными и обозначая Dc=k после интегрирования |
||
уравнения (1) получаем: x2 = 2kτ |
(2) |
|
т.е. в |
изотермических условиях |
квадрат толщины слоя продукта реакции |
пропорционален времени.
Поскольку измерить толщину слоя продукта реакции практически очень трудно, особенно в порошкообразных смесях, Яндер впоследствии видоизменил зависимость (2) и выразил ее через степень химического превращения компонента, покрываемого слоем продукта реакции, что достаточно легко определить обычными методами количественного анализа. Видоизмененная формула Яндера для изотермических условий имеет вид:
(3)
Где y -степень превращения покрываемого компонента, т.е. его количество, вошедшее в реакцию, мас.%; k -постоянная.
Применимость уравнения (3) для описания конкретного твердофазового процесса
проверяется с помощью |
графического построения зависимости величины (l - |
)2 от τ, |
которая должна быть прямой, проходящей через начало |
координат. Линейный ход этой зависимости указывает на правомерность описания кинетики твердофазовой реакции с помощью уравнения Яндера.
A.M. Гинстлинг и Б.И. Броунштейн рассмотрели кинетическую модель процесса твердофазового взаимодействия, учитывающую сферичность реагирующих зерен в смеси, и предложили следующее уравнение кинетики твердофазовых реакций для
изотермического процесса: |
|
1- 2/3G-(1-G)2/3 = k''τ, |
(4) |
где G — степень превращения покрываемого компонента в долях единицы; τ — время; k постоянная.
Уравнение (4) оказалось более строгим и для многих систем дает более точные результаты, особенно при больших степенях превращения компонентов и больших значениях τ, чем уравнения Яндера.
45

Следует отметить, что уравнения Яндера и Гинстлинга — Броунштейна, основанные на одном и том же исходном положении: скорость твердофазовой реакции обратно пропорциональна толщине слоя продукта реакции, были получены исходя из предположения об образовании продукта реакции путем односторонней диффузии покрывающего компонента в глубь зерен покрываемого компонента.
Поскольку в реальных условиях твердофазовые реакции могут осуществляться также за счет противодиффузии реагентов через слой продукта реакции, рассмотренные выше кинетические уравнения, не являются универсальными и удовлетворительно описывают кинетику отдельных твердофазовых реакций или их стадий.
Факторы, влияющие на скорость твердофазовых реакций 1. Температура.
Температура - один из важнейших факторов, влияющих на скорость твердофазовых реакций. Если процесс протекает в диффузионной области (а это справедливо для большинства реакций в силикатных системах), то скорость реакции определяется температурной зависимостью коэффициента диффузии:
где А - коэффициент, формально равный коэффициенту диффузии при Т=∞
Q- энергия активации процесса диффузии или энергия «разрыхления» решетки, зависящая от сил связи между ее структурными элементами;
R— газовая постоянная;
Т — абсолютная температура.
В соответствии с уравнением (5) величина коэффициента диффузии D и, следовательно, скорость твердофазовой реакции возрастают с увеличением температуры и уменьшением энергии активации
Следует подчеркнуть, что скорость диффузионного процесса, характеризующегося сравнительно небольшой энергией активации, изменяется с температурой примерно на 10—40% при ее изменении на 10°С, т.е. относительно медленно по сравнению с изменением скорости химической реакции (повышение температуры на 10°С увеличивает скорость химической реакции в 2—3 и более раза).
Было замечено, что процесс диффузии в кристаллической решетке различных веществ становится возможным при некоторой определенной температуре (так называемой температуре Таммана), которую для силикатов с очень грубым приближением можно принять равной 0,8Тпл, где Тпл — температура плавления вещества. Эту температуру. очень приближенно, можно считать минимальной температурой, при которой данное вещество с заметной скоростью вступает в твердофазовое взаимодействие. Следует, правда, учитывать возможность получения веществ в активном метастабильном состоянии, реакционная способность которых выше, чем у веществ в стабильном состоянии. В общем, твердофазовые реакции в большинстве силикатных систем с практически приемлемой скоростью протекают лишь при относительно высокой температуре (нередко выше 1000°С), хотя большинство реакций силикатообразования начинаются при относительно низких температурах (порядка 500...700°С).
2. Гранулометрический состав порошков. Твердофазовые реакции обычно протекают в порошкообразных смесях, в которых отдельные зерна соприкасаются между собой лишь небольшой частью поверхности. Так, в реальных порошкообразных массах
46
поверхность контакта между зернами составляет от 10-4 до 10-7 (в долях от общей поверхности). Поскольку чисто твердофазовые реакции (без участия жидкой и газообразной фаз) протекают именно на поверхности контакта зерен, то величина площади их соприкосновения очень сильно влияет на скорость подобных реакций. При прочих равных условиях скорость чисто твердофазового взаимодействия пропорциональна поверхности контакта реагирующих веществ и увеличение поверхности контакта между зернами способствует ускорению твердофазовых реакций.
Поскольку площадь контакта между зернами порошкообразных веществ увеличивается при уменьшении их размеров, дисперсность порошков оказывает существенное влияние на скорость твердофазовых реакций: с увеличением дисперсности скорость этих реакций при прочих равных условиях возрастает. Большое значение имеет также гранулометрический состав порошков: полидисперсные (многофракционные) порошки имеют меньшую пористость и большую поверхность контакта между зернами, что обусловлено заполнением мелкими зернами пространства между более крупными зернами. В таких порошках скорость твердофазового взаимодействия может быть существенно выше, чем в монодисперсных.
3.Давление. Важным фактором, влияющим на скорость твердофазовых реакций, является давление, прикладываемое к порошкообразным смесям, поскольку площадь поверхности контакта между зернами пропорциональна давлению сжатия. Увеличение поверхности контакта порошков при сжатии происходит за счет возрастания площади имеющихся контактных участков в результате пластической иди хрупкой деформации, а также за счет возникновения новых мест контактов. При использовании этого фактора речь может идти: а) о предварительном сжатии (прессовании) порошкообразной смеси перед осуществлением в ней реакции; б) о проведении самой реакции под давлением (горячее прессование). Прессование порошков может явиться эффективным способом повышения скорости твердофазовых реакций. Следует, однако, отметить, что при твердофазовой реакции с участием газовой или жидкой фазы повышение давления может не только не дать положительного эффекта, но даже уменьшить скорость реакции.
4.Присутствие газовой или жидкой фазы. Большое влияние на скорость твердофазовых реакций оказывает присутствие в системе реагирующих веществ газовой или жидкой фазы, которые сами непосредственно в реакции участия не принимают.
Как уже упоминалось, поверхность контакта между зернами в реальных порошках составляет лишь незначительную часть от их общей поверхности. Расчеты, проведенные с учетом указанного обстоятельства и возможных значений коэффициента диффузии в твердых телах, показывают, что если не принимать во внимание поверхностную диффузию, то степень превращения твердых тел за один час массопередачи путем внутренней диффузии не должна превышать 1 %. Однако в действительности скорость многих твердофазовых реакций, как правило, оказывается на 2—4 порядка выше, чем скорость, получаемая при подобных теоретических расчетах. Последнее показывает, что в случаях, наблюдаемых на практике, существенную роль играет поверхностная диффузия или массопередача, осуществляемая не через обычные контактные участки свободно насыпанного порошка, а преимущественно другим путем, например за счет перехода твердого тела в жидкое или газообразное состояние и его диффузии в жидкой или газовой фазе к поверхности другого тела. Экспериментальные данные показывают, что второй
47
путь часто реализуется и играет существенную роль во многих реальных твердофазовых процессах.
Значительное увеличение скорости твердофазовых реакций в присутствии газовой или жидкой фазы, не принимающих непосредственного участия в самой реакции, объясняется двумя факторами: а) увеличением площади реакционной поверхности, которая за счет омывания, обволакивания этими фазами твердых зерен приближается к общей площади поверхности зерен реагентов; б) значительно более высокой величиной коэффициента диффузии в газе и жидкости, в которые переходит, по крайней мере, один из реагирующих компонентов, по сравнению с твердой фазой.
Сказанное выше, естественно, не отрицает возможности протекания чисто твердофазовых реакций, но показывает, что в реальных условиях влияние газа и жидкости на реакции в смесях твердых веществ весьма велико и может быть использовано для интенсификации этих реакций в различных технологических процессах.
СПЕКАНИЕ
Сущность, движущая сила и виды спекания
Вобщем случае под спеканием понимается происходящий при высоких температурах процесс получения из слабосвязанного пористого зернистого материала плотного и прочного камневидного тела, сопровождающийся, как правило, уменьшением внешних размеров спекающегося тела (усадкой).
Вреальных условиях в технологии силикатных и других тугоплавких материалов спеканию подвергаются заготовки (сырец), сформованные различными методами из тонкозернистых масс. В зависимости от способа формования и гранулометрического состава исходного порошка такие заготовки имеют общую пористость порядка 25...60 об.
%(под порами понимается свободное пространство между отдельными зернами, слагающими заготовку, в также поры внутри зерен отдельных кристаллов). Конечным результатом спекания является получение из конгломерата слабосвязанных силами адгезии и трения плотного малопористого и прочного камневидного тела, свойства которого обусловлены силами химической связи.
Таким образом, спекание, в сущности, сводится к «удалению» пор из пористого тела за счет их самопроизвольного зарастания (заполнения веществом) при высокой температуре.
Степень спекания можно характеризовать относительной плотностью
ρотн=ρкаж/Dист или относительной пористостью Потн=П2/П1,
ρкаж _ кажущаяся плотность (отношение массы тела к его объему, включая объем пор);
Dист _ истинная плотность (отношение массы тела к его объему, исключая объем пор);
П1 и П2 — общая пористость, соответственно, до и после спекания.
При ρотн = 1 и Потн = 0 достигается абсолютное спекание материала, при котором его общая пористость становится равной нулю.
С термодинамической точки зрения спекание представляет собой
нестационарный необратимый процесс перехода системы в более стабильное, устойчивое состояние за счет самопроизвольного уплотнения дисперсного пористого тела. Движущей силой процесса спекания является поверхностная энергия. В исходном состоянии пористое тело имеет развитую внутреннюю межфазовую поверхность и представляет
48
собой систему, далекую от термодинамического равновесия. Это обусловлено повышенным запасом свободной поверхностной энергии. Как известно, любая система имеет тенденцию к сокращению межфазовой поверхности, что равносильно уменьшению поверхностной и, следовательно, общей энергии системы. При спекании эта тенденция реализуется за счет заполнения веществом пор между зернами и внутри зерен, что приводит к сокращению внутренней поверхности тела.
Основным физическим процессом при спекании является процесс массопереноса (диффузии), обеспечивающий заполнение пор веществом. В зависимости от механизма массопереноса различают несколько видов спекания: твердофазовое (диффузионное) спекание, жидкостное спекание, спекание за счет процесса испарение — конденсация, спекание за счет пластической деформации под давлением, реакционное спекание (за счет протекания химической реакции). Наиболее часто встречающимися в технологии силикатных и других тугоплавких материалов являются твердофазовое и жидкостное спекание. В зависимости от состава спекающегося материала и условий спекания один из его видов может преобладать, однако для многих материалов часто наблюдается совместное действие различных видов спекания. Ниже приведена характеристика отдельных видов спекания в их чистом виде.
Процесс спекания относится к одному из важнейших процессов, протекающих при изготовлении различных силикатных и других тугоплавких материалов и определяющих в значительной степени свойства получаемых продуктов. С увеличением степени спекания изделий возрастают их плотность, прочность, твердость, химическая стойкость, уменьшаются газо- и водопроницаемость.
Факторы, влияющие на процесс спекания
1. Температура и время спекания.
Температура является важнейшим фактором, влияющим на процесс спекания. При диффузионном твердофазовом спекании это влияние определяется сильной зависимостью от температуры процесса диффузии: увеличение температуры ускоряет диффузию и, следовательно, увеличивает скорость спекания и его степень. При жидкостном спекании ускоряющее влияние температуры на процесс спекания обусловлено уменьшением вязкости и улучшением смачивающей способности жидкой фазы при повышении температуры.
Скорость спекания изменяется во времени. Наиболее интенсивное спекание происходит до момента достижения пористости -10%, после чего обычно наблюдается уменьшение скорости уплотнения материала, одной из причин которого является увеличение давления газа в закрытых порах при уменьшении их размера, а также увеличение пути диффузии вакансий при твердофазовом спекании от поверхности внутренних пор к расположенной на значительном расстоянии от них границе зерен. Спекание также замедляет развивающийся при высоких температурах процесс рекристаллизации (см. ниже), т.е. процесс роста кристаллов.
Таким образом, скорость и степень спекания растут с увеличением температуры, а при данной температуре, если она достаточно велика, — с увеличением продолжительности выдержки. Последнее, однако, справедливо до известного предела, поскольку начиная с определенного момента времени скорость спекания резко замедляется. В связи с этим получение материала с возможно большей степенью спекания за счет увеличения времени спекания часто практически нецелесообразно.
2. Гранулометрический состав порошков.
49
На спекание очень большое влияние оказывают размер зерен в спекающемся теле и их распределение по размерам. Скорость и достигаемая степень спекания сильно возрастает при уменьшении размера зерен, в связи с чем, для интенсивного спекания требуется очень тонкое измельчение материала (вплоть до получения порошка с преобладающим размером зерен -1 мкм). Это объясняется рядом факторов:
увеличением начальной общей поверхности зернистого тела, что равносильно повышению свободной поверхностной энергии, т.е. движущей силы процесса спекания;
сокращением пути диффузии вакансий и атомов за счет уменьшения расстояния между источником и поглотителями вакансий;
увеличением числа контактов между зернами в единице объема, т.е. числа перемычек, перемещение которых приводит к заполнению пор материалом.
За счет повышения степени дисперсности одна и та же степень спекания в ряде случаев может быть достигнута при пониженных (на 200°С и более) температурах. Уменьшение размера зерен от 10 до 1 мкм приводит при прочих равных условиях к ускорению, например, жидкостного спекания в 10 раз.
Следует также отметить, что монодисперсный (содержащий зерна примерно одинакового размера) или близкий к нему порошок спекается медленнее и хуже, чем полидисперсный (содержащий зерна разных размеров), что объясняется меньшей пористостью и большей поверхностью взаимного контакта зерен в полидисперсных порошках.
3. Степень дефектности кристаллической решетки компонентов
спекающегося материала.
Применение исходных материалов в так называемом активном состоянии, т.е. имеющих далекую от равновесной, сильно искаженную решетку, позволяет в десятки и сотни (а иногда и более) раз повысить скорость спекания. Влияние этого фактора объясняется тем, что в материалах с сильно дефектной решеткой значительно возрастает коэффициент диффузии, т.е. ускоряется процесс массопереноса, который лежит в основе, например, механизма диффузионного спекания. На практике активность исходных материалов можно повысить различными способами. В частности, активность зависит от условий получения исходных материалов. Например, порошки, полученные термическим разложением солей при пониженных температурах, часто обладают искаженной (полуаморфной) решеткой и спекаются гораздо интенсивнее, чем те же материалы, полученные при более высоких температурах и имеющие более упорядоченную кристаллическую решетку. При интенсивном измельчении материала может происходить механическая активация. При этом увеличивается свободная энергия, уменьшаются пути диффузии, образуются аморфные слои, что в совокупности ускоряет спекание.
Дефектность кристаллической решетки может быть также повышена путем введения небольших добавок, образующих с основным веществом твердые растворы и вызывающих образование вакансий, за счет чего увеличивается коэффициент диффузии. Этот метод ускорения процесса спекания часто используется в технологии технической, особенно оксидной, керамики. Следует, однако, отметить, что влияние добавок на процесс спекания весьма сложно и не всегда однозначно. Образование плотных пленок примесей на поверхности зерен может, в некоторых случаях и замедлить процесс спекания. В
50