

- •Preface
- •Imaging Microscopic Features
- •Measuring the Crystal Structure
- •References
- •Contents
- •1.4 Simulating the Effects of Elastic Scattering: Monte Carlo Calculations
- •What Are the Main Features of the Beam Electron Interaction Volume?
- •How Does the Interaction Volume Change with Composition?
- •How Does the Interaction Volume Change with Incident Beam Energy?
- •How Does the Interaction Volume Change with Specimen Tilt?
- •1.5 A Range Equation To Estimate the Size of the Interaction Volume
- •References
- •2: Backscattered Electrons
- •2.1 Origin
- •2.2.1 BSE Response to Specimen Composition (η vs. Atomic Number, Z)
- •SEM Image Contrast with BSE: “Atomic Number Contrast”
- •SEM Image Contrast: “BSE Topographic Contrast—Number Effects”
- •2.2.3 Angular Distribution of Backscattering
- •Beam Incident at an Acute Angle to the Specimen Surface (Specimen Tilt > 0°)
- •SEM Image Contrast: “BSE Topographic Contrast—Trajectory Effects”
- •2.2.4 Spatial Distribution of Backscattering
- •Depth Distribution of Backscattering
- •Radial Distribution of Backscattered Electrons
- •2.3 Summary
- •References
- •3: Secondary Electrons
- •3.1 Origin
- •3.2 Energy Distribution
- •3.3 Escape Depth of Secondary Electrons
- •3.8 Spatial Characteristics of Secondary Electrons
- •References
- •4: X-Rays
- •4.1 Overview
- •4.2 Characteristic X-Rays
- •4.2.1 Origin
- •4.2.2 Fluorescence Yield
- •4.2.3 X-Ray Families
- •4.2.4 X-Ray Nomenclature
- •4.2.6 Characteristic X-Ray Intensity
- •Isolated Atoms
- •X-Ray Production in Thin Foils
- •X-Ray Intensity Emitted from Thick, Solid Specimens
- •4.3 X-Ray Continuum (bremsstrahlung)
- •4.3.1 X-Ray Continuum Intensity
- •4.3.3 Range of X-ray Production
- •4.4 X-Ray Absorption
- •4.5 X-Ray Fluorescence
- •References
- •5.1 Electron Beam Parameters
- •5.2 Electron Optical Parameters
- •5.2.1 Beam Energy
- •Landing Energy
- •5.2.2 Beam Diameter
- •5.2.3 Beam Current
- •5.2.4 Beam Current Density
- •5.2.5 Beam Convergence Angle, α
- •5.2.6 Beam Solid Angle
- •5.2.7 Electron Optical Brightness, β
- •Brightness Equation
- •5.2.8 Focus
- •Astigmatism
- •5.3 SEM Imaging Modes
- •5.3.1 High Depth-of-Field Mode
- •5.3.2 High-Current Mode
- •5.3.3 Resolution Mode
- •5.3.4 Low-Voltage Mode
- •5.4 Electron Detectors
- •5.4.1 Important Properties of BSE and SE for Detector Design and Operation
- •Abundance
- •Angular Distribution
- •Kinetic Energy Response
- •5.4.2 Detector Characteristics
- •Angular Measures for Electron Detectors
- •Elevation (Take-Off) Angle, ψ, and Azimuthal Angle, ζ
- •Solid Angle, Ω
- •Energy Response
- •Bandwidth
- •5.4.3 Common Types of Electron Detectors
- •Backscattered Electrons
- •Passive Detectors
- •Scintillation Detectors
- •Semiconductor BSE Detectors
- •5.4.4 Secondary Electron Detectors
- •Everhart–Thornley Detector
- •Through-the-Lens (TTL) Electron Detectors
- •TTL SE Detector
- •TTL BSE Detector
- •Measuring the DQE: BSE Semiconductor Detector
- •References
- •6: Image Formation
- •6.1 Image Construction by Scanning Action
- •6.2 Magnification
- •6.3 Making Dimensional Measurements With the SEM: How Big Is That Feature?
- •Using a Calibrated Structure in ImageJ-Fiji
- •6.4 Image Defects
- •6.4.1 Projection Distortion (Foreshortening)
- •6.4.2 Image Defocusing (Blurring)
- •6.5 Making Measurements on Surfaces With Arbitrary Topography: Stereomicroscopy
- •6.5.1 Qualitative Stereomicroscopy
- •Fixed beam, Specimen Position Altered
- •Fixed Specimen, Beam Incidence Angle Changed
- •6.5.2 Quantitative Stereomicroscopy
- •Measuring a Simple Vertical Displacement
- •References
- •7: SEM Image Interpretation
- •7.1 Information in SEM Images
- •7.2.2 Calculating Atomic Number Contrast
- •Establishing a Robust Light-Optical Analogy
- •Getting It Wrong: Breaking the Light-Optical Analogy of the Everhart–Thornley (Positive Bias) Detector
- •Deconstructing the SEM/E–T Image of Topography
- •SUM Mode (A + B)
- •DIFFERENCE Mode (A−B)
- •References
- •References
- •9: Image Defects
- •9.1 Charging
- •9.1.1 What Is Specimen Charging?
- •9.1.3 Techniques to Control Charging Artifacts (High Vacuum Instruments)
- •Observing Uncoated Specimens
- •Coating an Insulating Specimen for Charge Dissipation
- •Choosing the Coating for Imaging Morphology
- •9.2 Radiation Damage
- •9.3 Contamination
- •References
- •10: High Resolution Imaging
- •10.2 Instrumentation Considerations
- •10.4.1 SE Range Effects Produce Bright Edges (Isolated Edges)
- •10.4.4 Too Much of a Good Thing: The Bright Edge Effect Hinders Locating the True Position of an Edge for Critical Dimension Metrology
- •10.5.1 Beam Energy Strategies
- •Low Beam Energy Strategy
- •High Beam Energy Strategy
- •Making More SE1: Apply a Thin High-δ Metal Coating
- •Making Fewer BSEs, SE2, and SE3 by Eliminating Bulk Scattering From the Substrate
- •10.6 Factors That Hinder Achieving High Resolution
- •10.6.2 Pathological Specimen Behavior
- •Contamination
- •Instabilities
- •References
- •11: Low Beam Energy SEM
- •11.3 Selecting the Beam Energy to Control the Spatial Sampling of Imaging Signals
- •11.3.1 Low Beam Energy for High Lateral Resolution SEM
- •11.3.2 Low Beam Energy for High Depth Resolution SEM
- •11.3.3 Extremely Low Beam Energy Imaging
- •References
- •12.1.1 Stable Electron Source Operation
- •12.1.2 Maintaining Beam Integrity
- •12.1.4 Minimizing Contamination
- •12.3.1 Control of Specimen Charging
- •12.5 VPSEM Image Resolution
- •References
- •13: ImageJ and Fiji
- •13.1 The ImageJ Universe
- •13.2 Fiji
- •13.3 Plugins
- •13.4 Where to Learn More
- •References
- •14: SEM Imaging Checklist
- •14.1.1 Conducting or Semiconducting Specimens
- •14.1.2 Insulating Specimens
- •14.2 Electron Signals Available
- •14.2.1 Beam Electron Range
- •14.2.2 Backscattered Electrons
- •14.2.3 Secondary Electrons
- •14.3 Selecting the Electron Detector
- •14.3.2 Backscattered Electron Detectors
- •14.3.3 “Through-the-Lens” Detectors
- •14.4 Selecting the Beam Energy for SEM Imaging
- •14.4.4 High Resolution SEM Imaging
- •Strategy 1
- •Strategy 2
- •14.5 Selecting the Beam Current
- •14.5.1 High Resolution Imaging
- •14.5.2 Low Contrast Features Require High Beam Current and/or Long Frame Time to Establish Visibility
- •14.6 Image Presentation
- •14.6.1 “Live” Display Adjustments
- •14.6.2 Post-Collection Processing
- •14.7 Image Interpretation
- •14.7.1 Observer’s Point of View
- •14.7.3 Contrast Encoding
- •14.8.1 VPSEM Advantages
- •14.8.2 VPSEM Disadvantages
- •15: SEM Case Studies
- •15.1 Case Study: How High Is That Feature Relative to Another?
- •15.2 Revealing Shallow Surface Relief
- •16.1.2 Minor Artifacts: The Si-Escape Peak
- •16.1.3 Minor Artifacts: Coincidence Peaks
- •16.1.4 Minor Artifacts: Si Absorption Edge and Si Internal Fluorescence Peak
- •16.2 “Best Practices” for Electron-Excited EDS Operation
- •16.2.1 Operation of the EDS System
- •Choosing the EDS Time Constant (Resolution and Throughput)
- •Choosing the Solid Angle of the EDS
- •Selecting a Beam Current for an Acceptable Level of System Dead-Time
- •16.3.1 Detector Geometry
- •16.3.2 Process Time
- •16.3.3 Optimal Working Distance
- •16.3.4 Detector Orientation
- •16.3.5 Count Rate Linearity
- •16.3.6 Energy Calibration Linearity
- •16.3.7 Other Items
- •16.3.8 Setting Up a Quality Control Program
- •Using the QC Tools Within DTSA-II
- •Creating a QC Project
- •Linearity of Output Count Rate with Live-Time Dose
- •Resolution and Peak Position Stability with Count Rate
- •Solid Angle for Low X-ray Flux
- •Maximizing Throughput at Moderate Resolution
- •References
- •17: DTSA-II EDS Software
- •17.1 Getting Started With NIST DTSA-II
- •17.1.1 Motivation
- •17.1.2 Platform
- •17.1.3 Overview
- •17.1.4 Design
- •Simulation
- •Quantification
- •Experiment Design
- •Modeled Detectors (. Fig. 17.1)
- •Window Type (. Fig. 17.2)
- •The Optimal Working Distance (. Figs. 17.3 and 17.4)
- •Elevation Angle
- •Sample-to-Detector Distance
- •Detector Area
- •Crystal Thickness
- •Number of Channels, Energy Scale, and Zero Offset
- •Resolution at Mn Kα (Approximate)
- •Azimuthal Angle
- •Gold Layer, Aluminum Layer, Nickel Layer
- •Dead Layer
- •Zero Strobe Discriminator (. Figs. 17.7 and 17.8)
- •Material Editor Dialog (. Figs. 17.9, 17.10, 17.11, 17.12, 17.13, and 17.14)
- •17.2.1 Introduction
- •17.2.2 Monte Carlo Simulation
- •17.2.4 Optional Tables
- •References
- •18: Qualitative Elemental Analysis by Energy Dispersive X-Ray Spectrometry
- •18.1 Quality Assurance Issues for Qualitative Analysis: EDS Calibration
- •18.2 Principles of Qualitative EDS Analysis
- •Exciting Characteristic X-Rays
- •Fluorescence Yield
- •X-ray Absorption
- •Si Escape Peak
- •Coincidence Peaks
- •18.3 Performing Manual Qualitative Analysis
- •Beam Energy
- •Choosing the EDS Resolution (Detector Time Constant)
- •Obtaining Adequate Counts
- •18.4.1 Employ the Available Software Tools
- •18.4.3 Lower Photon Energy Region
- •18.4.5 Checking Your Work
- •18.5 A Worked Example of Manual Peak Identification
- •References
- •19.1 What Is a k-ratio?
- •19.3 Sets of k-ratios
- •19.5 The Analytical Total
- •19.6 Normalization
- •19.7.1 Oxygen by Assumed Stoichiometry
- •19.7.3 Element by Difference
- •19.8 Ways of Reporting Composition
- •19.8.1 Mass Fraction
- •19.8.2 Atomic Fraction
- •19.8.3 Stoichiometry
- •19.8.4 Oxide Fractions
- •Example Calculations
- •19.9 The Accuracy of Quantitative Electron-Excited X-ray Microanalysis
- •19.9.1 Standards-Based k-ratio Protocol
- •19.9.2 “Standardless Analysis”
- •19.10 Appendix
- •19.10.1 The Need for Matrix Corrections To Achieve Quantitative Analysis
- •19.10.2 The Physical Origin of Matrix Effects
- •19.10.3 ZAF Factors in Microanalysis
- •X-ray Generation With Depth, φ(ρz)
- •X-ray Absorption Effect, A
- •X-ray Fluorescence, F
- •References
- •20.2 Instrumentation Requirements
- •20.2.1 Choosing the EDS Parameters
- •EDS Spectrum Channel Energy Width and Spectrum Energy Span
- •EDS Time Constant (Resolution and Throughput)
- •EDS Calibration
- •EDS Solid Angle
- •20.2.2 Choosing the Beam Energy, E0
- •20.2.3 Measuring the Beam Current
- •20.2.4 Choosing the Beam Current
- •Optimizing Analysis Strategy
- •20.3.4 Ba-Ti Interference in BaTiSi3O9
- •20.4 The Need for an Iterative Qualitative and Quantitative Analysis Strategy
- •20.4.2 Analysis of a Stainless Steel
- •20.5 Is the Specimen Homogeneous?
- •20.6 Beam-Sensitive Specimens
- •20.6.1 Alkali Element Migration
- •20.6.2 Materials Subject to Mass Loss During Electron Bombardment—the Marshall-Hall Method
- •Thin Section Analysis
- •Bulk Biological and Organic Specimens
- •References
- •21: Trace Analysis by SEM/EDS
- •21.1 Limits of Detection for SEM/EDS Microanalysis
- •21.2.1 Estimating CDL from a Trace or Minor Constituent from Measuring a Known Standard
- •21.2.2 Estimating CDL After Determination of a Minor or Trace Constituent with Severe Peak Interference from a Major Constituent
- •21.3 Measurements of Trace Constituents by Electron-Excited Energy Dispersive X-ray Spectrometry
- •The Inevitable Physics of Remote Excitation Within the Specimen: Secondary Fluorescence Beyond the Electron Interaction Volume
- •Simulation of Long-Range Secondary X-ray Fluorescence
- •NIST DTSA II Simulation: Vertical Interface Between Two Regions of Different Composition in a Flat Bulk Target
- •NIST DTSA II Simulation: Cubic Particle Embedded in a Bulk Matrix
- •21.5 Summary
- •References
- •22.1.2 Low Beam Energy Analysis Range
- •22.2 Advantage of Low Beam Energy X-Ray Microanalysis
- •22.2.1 Improved Spatial Resolution
- •22.3 Challenges and Limitations of Low Beam Energy X-Ray Microanalysis
- •22.3.1 Reduced Access to Elements
- •22.3.3 At Low Beam Energy, Almost Everything Is Found To Be Layered
- •Analysis of Surface Contamination
- •References
- •23: Analysis of Specimens with Special Geometry: Irregular Bulk Objects and Particles
- •23.2.1 No Chemical Etching
- •23.3 Consequences of Attempting Analysis of Bulk Materials With Rough Surfaces
- •23.4.1 The Raw Analytical Total
- •23.4.2 The Shape of the EDS Spectrum
- •23.5 Best Practices for Analysis of Rough Bulk Samples
- •23.6 Particle Analysis
- •Particle Sample Preparation: Bulk Substrate
- •The Importance of Beam Placement
- •Overscanning
- •“Particle Mass Effect”
- •“Particle Absorption Effect”
- •The Analytical Total Reveals the Impact of Particle Effects
- •Does Overscanning Help?
- •23.6.6 Peak-to-Background (P/B) Method
- •Specimen Geometry Severely Affects the k-ratio, but Not the P/B
- •Using the P/B Correspondence
- •23.7 Summary
- •References
- •24: Compositional Mapping
- •24.2 X-Ray Spectrum Imaging
- •24.2.1 Utilizing XSI Datacubes
- •24.2.2 Derived Spectra
- •SUM Spectrum
- •MAXIMUM PIXEL Spectrum
- •24.3 Quantitative Compositional Mapping
- •24.4 Strategy for XSI Elemental Mapping Data Collection
- •24.4.1 Choosing the EDS Dead-Time
- •24.4.2 Choosing the Pixel Density
- •24.4.3 Choosing the Pixel Dwell Time
- •“Flash Mapping”
- •High Count Mapping
- •References
- •25.1 Gas Scattering Effects in the VPSEM
- •25.1.1 Why Doesn’t the EDS Collimator Exclude the Remote Skirt X-Rays?
- •25.2 What Can Be Done To Minimize gas Scattering in VPSEM?
- •25.2.2 Favorable Sample Characteristics
- •Particle Analysis
- •25.2.3 Unfavorable Sample Characteristics
- •References
- •26.1 Instrumentation
- •26.1.2 EDS Detector
- •26.1.3 Probe Current Measurement Device
- •Direct Measurement: Using a Faraday Cup and Picoammeter
- •A Faraday Cup
- •Electrically Isolated Stage
- •Indirect Measurement: Using a Calibration Spectrum
- •26.1.4 Conductive Coating
- •26.2 Sample Preparation
- •26.2.1 Standard Materials
- •26.2.2 Peak Reference Materials
- •26.3 Initial Set-Up
- •26.3.1 Calibrating the EDS Detector
- •Selecting a Pulse Process Time Constant
- •Energy Calibration
- •Quality Control
- •Sample Orientation
- •Detector Position
- •Probe Current
- •26.4 Collecting Data
- •26.4.1 Exploratory Spectrum
- •26.4.2 Experiment Optimization
- •26.4.3 Selecting Standards
- •26.4.4 Reference Spectra
- •26.4.5 Collecting Standards
- •26.4.6 Collecting Peak-Fitting References
- •26.5 Data Analysis
- •26.5.2 Quantification
- •26.6 Quality Check
- •Reference
- •27.2 Case Study: Aluminum Wire Failures in Residential Wiring
- •References
- •28: Cathodoluminescence
- •28.1 Origin
- •28.2 Measuring Cathodoluminescence
- •28.3 Applications of CL
- •28.3.1 Geology
- •Carbonado Diamond
- •Ancient Impact Zircons
- •28.3.2 Materials Science
- •Semiconductors
- •Lead-Acid Battery Plate Reactions
- •28.3.3 Organic Compounds
- •References
- •29.1.1 Single Crystals
- •29.1.2 Polycrystalline Materials
- •29.1.3 Conditions for Detecting Electron Channeling Contrast
- •Specimen Preparation
- •Instrument Conditions
- •29.2.1 Origin of EBSD Patterns
- •29.2.2 Cameras for EBSD Pattern Detection
- •29.2.3 EBSD Spatial Resolution
- •29.2.5 Steps in Typical EBSD Measurements
- •Sample Preparation for EBSD
- •Align Sample in the SEM
- •Check for EBSD Patterns
- •Adjust SEM and Select EBSD Map Parameters
- •Run the Automated Map
- •29.2.6 Display of the Acquired Data
- •29.2.7 Other Map Components
- •29.2.10 Application Example
- •Application of EBSD To Understand Meteorite Formation
- •29.2.11 Summary
- •Specimen Considerations
- •EBSD Detector
- •Selection of Candidate Crystallographic Phases
- •Microscope Operating Conditions and Pattern Optimization
- •Selection of EBSD Acquisition Parameters
- •Collect the Orientation Map
- •References
- •30.1 Introduction
- •30.2 Ion–Solid Interactions
- •30.3 Focused Ion Beam Systems
- •30.5 Preparation of Samples for SEM
- •30.5.1 Cross-Section Preparation
- •30.5.2 FIB Sample Preparation for 3D Techniques and Imaging
- •30.6 Summary
- •References
- •31: Ion Beam Microscopy
- •31.1 What Is So Useful About Ions?
- •31.2 Generating Ion Beams
- •31.3 Signal Generation in the HIM
- •31.5 Patterning with Ion Beams
- •31.7 Chemical Microanalysis with Ion Beams
- •References
- •Appendix
- •A Database of Electron–Solid Interactions
- •A Database of Electron–Solid Interactions
- •Introduction
- •Backscattered Electrons
- •Secondary Yields
- •Stopping Powers
- •X-ray Ionization Cross Sections
- •Conclusions
- •References
- •Index
- •Reference List
- •Index
\334 Chapter 20 · Quantitative Analysis: The SEM/EDS Elemental Microanalysis k-ratio Procedure for Bulk Specimens, Step-by-Step
Counts
Counts
160 000
140 000
120 000
10 000
80 000
60 000
40 000
20 000
0
0.0
20 000
15 000
10 000
5 000
0 2.5
Corning Glass A E0 = 15 keV 100 µm square 50 µm square 20 µm square 5 µm square
2 µm square
1 µm square spot
CorningA_1mu_15kV15nA
CorningA_2mu_15kV15nA
CorningA_5mu_15kV15nA
CorningA_20mu_15kV15nA
CorningA_50mu_15kV15nA
CorningA_100mu_15kV15nA
CorningA_spot_15kV15nA
0.2 |
0.4 |
0.6 |
0.8 |
1.0 |
1.2 |
1.4 |
1.6 |
1.8 |
2.0 |
|
|
|
|
Photon energy (keV) |
|
|
|
|
|
|
Corning Glass A |
|
|
|
|
|
CorningA_1mu_15kV15nA |
|
|
|
E0 = 15 keV |
|
|
|
|
|
|
CorningA_2mu_15kV15nA |
|
|
|
|
|
|
|
|
CorningA_5mu_15kV15nA |
|
|
|
100 µm square |
|
|
|
|
|
|
CorningA_20mu_15kV15nA |
|
|
|
|
|
|
|
|
CorningA_50mu_15kV15nA |
||
|
50 µm square |
|
|
|
|
|
|
CorningA_100mu_15kV15nA |
|
|
|
|
|
|
|
|
CorningA_spot_15kV15nA |
|
|
20 µm square
5 µm square
2 µm square
1 µm square spot
2.7 |
2.9 |
3.1 |
3.3 |
3.5 |
3.7 |
3.9 |
4.1 |
4.3 |
Photon energy (keV)
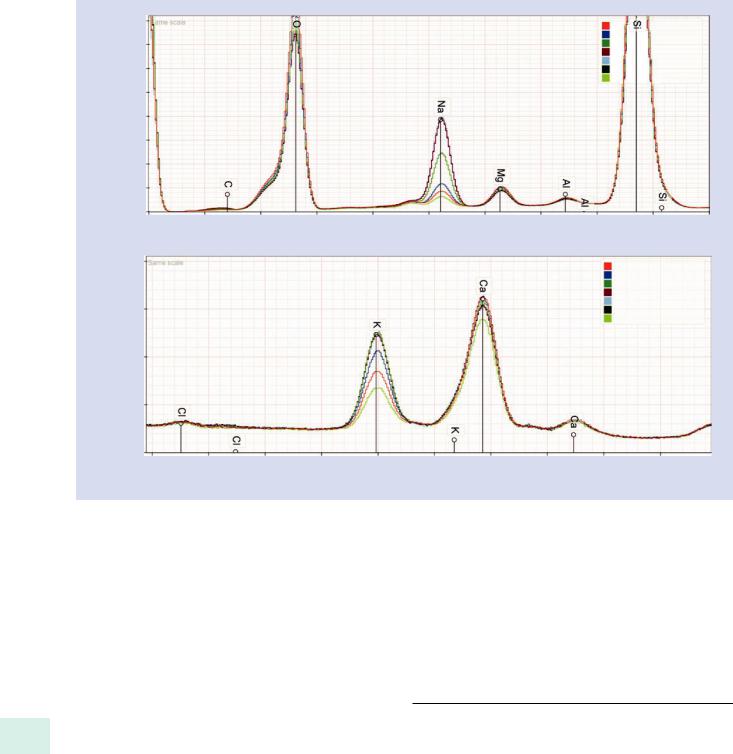
. Fig. 20.16 Corning glass A, showing Na and K migration as a function of dose for scanning beams covering various areas (20 keV, 10 nA)
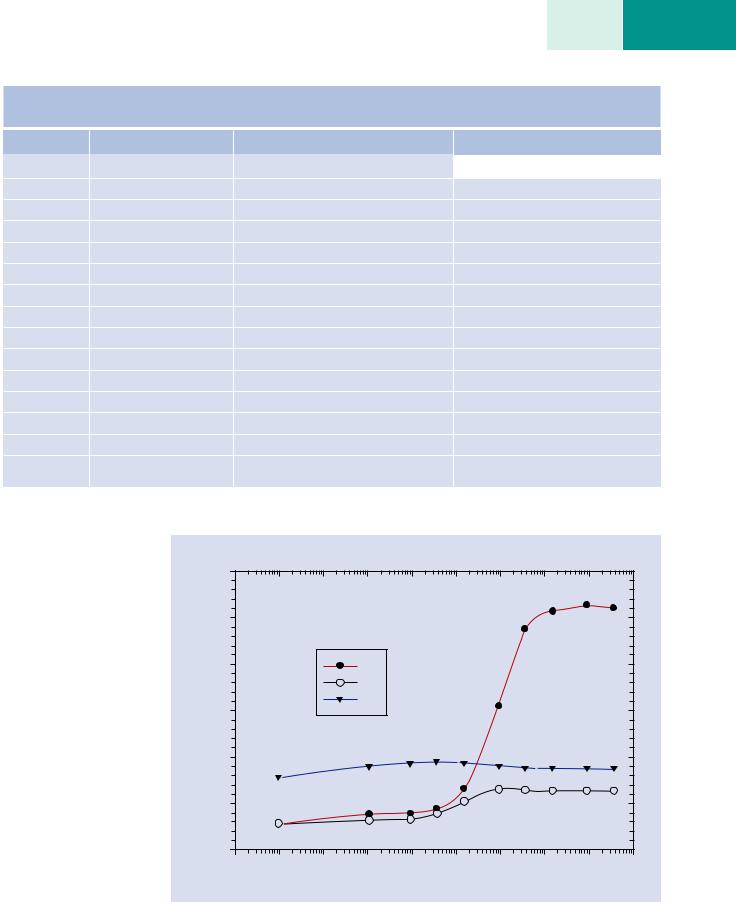
values for the glass, including the alkali elements Na and K, whereas the point beam results show reductions in the Na and K concentrations. . Figure 20.17 shows that the measured sodium and potassium concentrations increase to reach the synthesized values as the scanned area dimensions are increased to cover areas above 20 x 20-μm (nominal magnification 5 kX) for the particular dose utilized (15 keV, 1500 nA-s). Thus, while scanning a large homogeneous area obviously concedes the spatial resolution capability of electron-excited X-ray microanalysis, this approach may be the most expedient technique to control and minimize alkali element migration.
20 Materials that can serve as useful standards for sodium include certain crystalline minerals such as albite (NaAlSi3O8) in which the sodium is much more stable under electron bombardment. However, even for albite the use of a stationary high intensity point beam may produce significant migration effects, as shown in . Fig. 20.18 for spectra collected with a stationary point beam as a function of dose (upper)
and at the same dose with a fixed beam and two different sizes of scanned areas (lower). Thus, the use of a scanned area rather than a fixed beam may be necessary when collecting a standard spectrum, even on a crystalline material.
20.6.2\ Materials Subject to Mass Loss During Electron Bombardment—the Marshall-Hall Method
Thin Section Analysis
The X-ray microanalysis of biological and polymeric specimens is made difficult, and sometimes impossible, by several forms of radiation damage that are directly caused by the electron beam. At the beam energies used in the SEM (0.1–
30 keV), it is possible for the kinetic energy of individual beam electrons to break and/or rearrange chemical bonds. The radiation damage can release smaller molecules such as
335 |
20 |
20.6 · Beam-Sensitive Specimens
. Table 20.14 DTSA-II quantitative analysis of Corning glass A: Comparison of results with a fixed beam and scanned beam (100 μm
square) (15 keV/15 nA); oxygen by assumed stoichiometry
Element |
As-synthesized mass conc |
1500 nA-s (fixed beam) raw mass conc |
1500 nA-s (100 -μm2 scan) raw mass conc |
O |
0.4421 |
0.4644 ± 0.0009 |
0.4577 ± 0.0006 |
|
|
|
|
Na |
0.1061 |
0.0098 ± 0.0003 |
0.1076 ± 0.0004 |
Mg |
0.0160 |
0.0186 ± 0.0002 |
0.0164 ± 0.0001 |
Al |
0.0529 |
0.0058 ± 0.0001 |
0.0056 ± 0.0000 |
Si |
0.3111 |
0.3574 ± 0.0007 |
0.3239 ± 0.0005 |
K |
0.0238 |
0.0166 ± 0.0003 |
0.0257 ± 0.0002 |
Ca |
0.0359 |
0.0386 ± 0.0003 |
0.0350 ± 0.0001 |
Ti |
0.00474 |
0.0057 ± 0.0002 |
0.0051 ± 0.0001 |
Mn |
0.00775 |
0.0101 ± 0.0003 |
0.0082 ± 0.0001 |
Fe |
0.00762 |
0.0090 ± 0.0003 |
0.0077 ± 0.0001 |
Cu |
0.00935 |
0.0108 ± 0.0005 |
0.0096 ± 0.0003 |
Sn |
0.00150 |
0.0054 ± 0.0007 |
0.0045 ± 0.0003 |
Sb |
0.0146 |
0.0140 ± 0.0007 |
0.0125 ± 0.0002 |
Ba |
0.0050 |
0.0042 ± 0.0005 |
0.0042 ± 0.0002 |
Raw total |
|
0.9718 |
1.0254 |
. Fig. 20.17 Results of quantitative analysis of Corning glass A as a function of the size of the area scanned. Nominal magnifications indicated
Concentration (mass)
Corning glass A (E0 = 15 keV; 15 nA)
0.12
|
|
|
|
|
|
|
5 kX |
2 kX |
1 kX |
|
|
|
|
|
|
|
|
||
0.10 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
10 kX |
|
|
0.08 |
|
|
Na |
|
|
|
|
|
|
|
|
|
K |
|
|
|
|
|
|
0.06 |
|
|
Ca |
|
|
20 kX |
|
|
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
0.04 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
50 kX |
|
|
|
0.02 |
Fixed |
|
500 kX |
100 kX |
|
|
|
|
|
Beam |
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
200 kX |
|
|
|
|
|
0.00 |
|
|
|
|
|
|
|
|
|
1e-5 |
1e-4 |
1e-3 |
1e-2 |
1e-1 |
1e+0 |
1e+1 |
1e+2 |
1e+3 |
1e+4 |
Area bombarded (mm2)
\336 Chapter 20 · Quantitative Analysis: The SEM/EDS Elemental Microanalysis k-ratio Procedure for Bulk Specimens, Step-by-Step
Counts
Counts
7 000 |
|
|
|
Albite |
|
|
|
|
Albite_Point_10s_15kV15nA |
|
|
|
|
|
|
|
|
|
Albite_Point_50s_15kV15nA |
|
|
|
|
|
|
|
|
|
|
|
|
|
6 000 |
|
|
|
E0 = 15 keV |
|
|
|
|
Albite_Point_100s_15kV15nA |
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
Fixed beam |
|
|
|
|
|
|
5 000 |
|
|
|
10 s spectra |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
4 000 |
|
|
|
1st (10 s) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
5th (50 s) |
|
|
|
|
|
|
3 000 |
|
|
|
10th (100 s) |
|
|
|
|
|
|
2 000 |
|
|
|
|
|
|
|
|
|
|
1 000 |
|
|
|
|
|
|
|
|
|
|
0 |
0.2 |
0.4 |
0.6 |
0.8 |
1.0 |
1.2 |
1.4 |
1.6 |
1.8 |
2.0 |
0.0 |
||||||||||
|
|
|
|
|
Photon energy (keV) |
|
|
|
|
|
140 000 |
|
|
|
|
|
|
|
|
Albite_100kX_15kV15nA |
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
Albite |
|
|
|
|
Albite_10kX_15kV15nA |
|
120 000 |
|
|
|
|
|
|
|
Albite_spot_15kV15nA |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
E0 = 15 keV |
|
|
|
|
|
|
100 000 |
|
|
|
100 s spectra |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
80 000 |
|
|
|
1 µm x 1 µm |
|
|
|
|
|
|
|
|
|
10 µm x 10 µm |
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
||
60 000 |
|
|
|
Fixed beam |
|
|
|
|
|
|
40 000 |
|
|
|
|
|
|
|
|
|
|
20 000 |
|
|
|
|
|
|
|
|
|
|
0 |
0.2 |
0.4 |
0.6 |
0.8 |
1.0 |
1.2 |
1.4 |
1.6 |
1.8 |
2.0 |
0.0 |
||||||||||
Photon energy (keV)
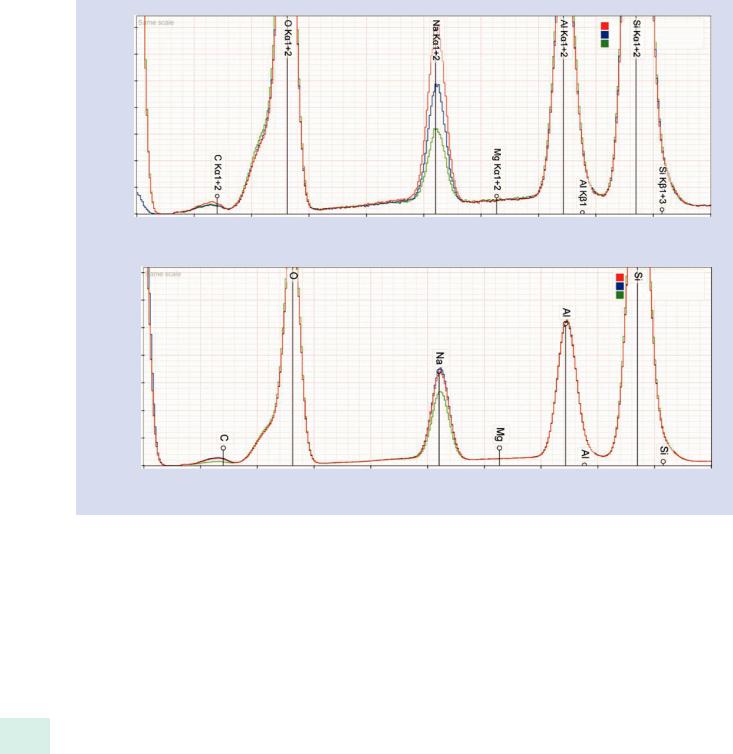
. Fig. 20.18 Albite (NaAlSi3O8); E0 = 15 keV, 15 nA: (upper) effect of increasing dose on the Na peak; (lower) effect of fixed beam versus scanned beam on the Na peak
CO, CO2, and H2O that evaporate into the vacuum, causing substantial mass loss from the interaction volume. At the highest beam currents, typically 10–100 nA, used with a focused beam at a static location, it is also possible to cause highly damaging temperature elevations, which further exacerbate mass loss. Indeed, when analyzing this specimen class it should be assumed that significant mass loss will occur during the measurement at each point of the specimen. If all constituents were lost at the same rate, then simply normalizing
20 the result would compensate for the mass loss that occurs duringtheaccumulationoftheX-rayspectrum.Unfortunately, the matrix constituents (principally carbon compounds and water) can be selectively lost, while the heavy elements of interest in biological microanalysis (e. g., Mg, P, S, K, Ca, Fe, etc.) remain in the bombarded region of the specimen and appear to be present at effectively higher concentration than existed in the original specimen. What is then required of any analytical procedure for biological and polymeric specimens is a mechanism to provide a meaningful analysis under these conditions of a specimen that undergoes continuous change.
Marshall and Hall (1966) and Hall (1968) made the original suggestion that the X-ray continuum could serve as an internal standard to monitor specimen changes. This assumption permitted development of the key procedure for beam-sensi- tive specimens that is used extensively in the biological community and that is also applicable in many types of polymer analysis. This application marks the earliest use of the X-ray continuum as a tool (rather than simply a hindrance) for analysis, and that work forms the basis for the development of the peak-to-local background method applied to challenging geometric forms such as particles and rough surfaces. The technique was initially developed for applications in the high beam current EPMA, but the procedure works well in the
SEM environment.
The Marshall–Hall method (Marshall and Hall 1966) requires that several key conditions:
\1.\ The specimen must be in the form of a thin section, where the condition of “thin” is satisfied when the incident beam penetrates with negligible energy loss. For an analytical beam energy of 10–30 keV, the energy loss

337 |
20 |
20.6 · Beam-Sensitive Specimens
passing through a section consisting of carbon approximately 100–200 nm in thickness will be less than
500 eV. This condition permits the beam energy to be treated as a constant, which is critical for the development of the correction formula. Biological specimens are thus usually analyzed in the form of thin sections cut to approximately 100-nm thickness by microtome. Polymers may also be analyzed when similarly prepared as thin sections by microtoming or by ion beam milling. Such a specimen configuration also has a distinct advantage for improving the spatial resolution of the analysis compared to a bulk specimen. The analytical volume in such thin specimens is approximately the cylinder defined by the incident beam diameter and the section thickness, which is at least a factor of 10–100 smaller in linear dimensions than the equivalent bulk specimen case at the same energy, as shown in the polymer etching experiment in the Interaction Volume module.
\2.\ The matrix composition must be dominated by light elements, for example, C, H, N, O, whose contributions will form nearly all of the X-ray continuum and whose concentrations are reasonably well known for the specimen. Elements of analytical interest such as Mg, P, S, Cl, K, Ca, and so on, the concentrations of which are unknown in the specimen, must only be present preferably as trace constituents (<0.01 mass fraction) so that their effect on the X-ray continuum can be neglected. When the concentration rises above the low end of the minor constituent range (e.g., 0.01 to 0.05 mass fraction or more), the analyte contribution to the continuum can no longer be ignored.
\3.\ A standard must be available with a known concentration of the trace/minor analyte of interest and for which the complete composition of low-atomic-number elements is also known and which is stable under electron beam bombardment. Glasses synthesized with low atomic number oxides such as boron oxide are suitable for this role. The closer the low–atomic-number element composition of the standard is to that of the unknown, the more accurate will be the results.
The detailed derivation yields the following general expression for the Marshall–Hall method:
CA
Ich |
= c |
|
|
|
|
|
AA |
|
|
(20.9) |
Icm |
|
|
|
Z 2 |
|
|
E |
|
||
|
|
|
||||||||
|
|
∑ Ci |
i |
loge 1.166 |
0 |
|
|
|||
|
Ai |
|
|
|||||||
|
|
i |
|
|
|
|
Ji |
\ |
||
|
|
|
|
|
|
|
|
|
|
|
In this equation, Ich is the characteristic intensity of the peak
of interest, for example, S K-L2,3 or Ca K-L2,3, and Icm is the continuum intensity of a continuum window of width E
placed somewhere in the high energy portion of the spectrum, typically above 8 keV, so that absorption effects are negligible and only mass effects are important. Ci is the mass
concentration, Zi is the atomic number, and Ai is the atomic weight. The subscript “A” identifies a specific trace or minor analyte of interest (e.g., Mg, P, S, Cl, Ca, Fe, etc.) in the organic matrix, while the subscript “i” represents all elements in the electron-excited region. E0 is the incident beam energy and J is the mean ionization energy, a function only of atomic number as used in the Bethe continuous energy loss equation
Assumption 2 provides that the quantity ∑(Ci•Zi2/Ai) in Eq. (20.9) for the biological or polymeric specimen to be analyzed is dominated by the low-Z constituents of the matrix. (Some representative values of ∑(Ci•Zi2/Ai) are 3.67 (water), 3.01 (nylon), 3.08 (polycarbonate) and 3.28 (protein with S). Typically the range is between 2.8 and 3.8 for most biological and many polymeric materials.) The unknown contribution of the analyte, CA, to the sum may be neglected when considering the specimen because CA is low when the analytes are trace constituents.
To perform a quantitative analysis, Eq. (20.9) is used in the following manner: A standard for which all elemental concentrations are known and which contains the analyte(s) of interest “A” is prepared as a thin cross section (satisfying assumption 3). This standard is measured under defined beam and spectrometer parameters to yield a characteristic- to-continuum ratio, IA/Icm. This measured ratio IA/Icm is set equal to the right side of Eq. (20.9). Since the target being irradiated is a reference standard, the atomic numbers Zi, atomic weights Ai and weight fractions Ci are known for all constituents, and the Ji values can be calculated as needed. The only unknown term is then the constant “c” in Eq. (20.9), which can now be determined by dividing the measured intensity ratio, IA/Icm, by the calculated term. Next, under the same measurement conditions, the characteristic “A” intensity and the continuum intensity at the chosen energy are determined for the specimen location(s). Providing that the low-Z elements that form the matrix of the specimen are similar to the standard, or in the optimum case these concentrations are actually known for the specimen (or can be estimated from other information about the actual, localized, material being irradiated by the electrons, and not some bulk property), then this value of “c” determined from the standard can be used to calculate the weight fraction of the analyte, CA, for the specimen.
This basic theme can be extended and several analytes— “A,” “B,” “C,” etc.—can be analyzed simultaneously if a suitable standard or suite of standards containing the analytes is available. The method can be extended to higher concentrations, but the details of this extension are beyond the scope of this book; a full description and derivation can be found in Kitazawa et al. (1983). Commercial computer X-ray analyzer systems may have the Marshall–Hall procedure included in their suite of analysis tools. The Marshall–Hall procedure works well for thin specimens in the “conventional” analytical energy regime (E0 ≥10 keV) of the SEM. The method will not work for specimens where the average atomic number is expected to vary significantly from one analysis point to another, or relative to that of the standard. A bulk specimen where the beam-damaged region is not constrained by the